
Vous êtes ici
Thématique:
Diagnostic étiologique d’une déficience intellectuelle
V. des Portes1, A. Verloes2, 4, D. Héron3, 4 et le réseau DéfiScience** pour la SFNP
1Centre de référence « Déficiences intellectuelles de causes rares », HFME, Hospices Civils de Lyon et Université Lyon 1, 59 boulevard Pinel, 69677 Bron, France 2Département de Génétique, INSERM U676, CHU Robert- Debré, AP- HP, 48 boulevard Serrurier, 75019 Paris, France 3Département de Génétique, INSERM CRicm, UMR- S975, Groupe Hospitalier Pitié- Salpêtrière, AP-HP, 47 boulevard Hôpital, 75013 Paris, France 4Centre de référence Maladies Rares « Déficiences intellectuelles de causes rares », Groupe Hospitalier Pitié- Salpêtrière, AP-HP, 47 boulevard Hôpital, 75013 Paris, France Auteur correspondant - Adresse e-mail :vincent.desportes@chu-lyon.fr (V. des Portes) ** Réseau des Centre de compétences associés aux Centres de référence Maladies Rares "déficiences intellectuelles de causes rares" (Paris et Lyon) et "malformations cérébelleuses" (Paris)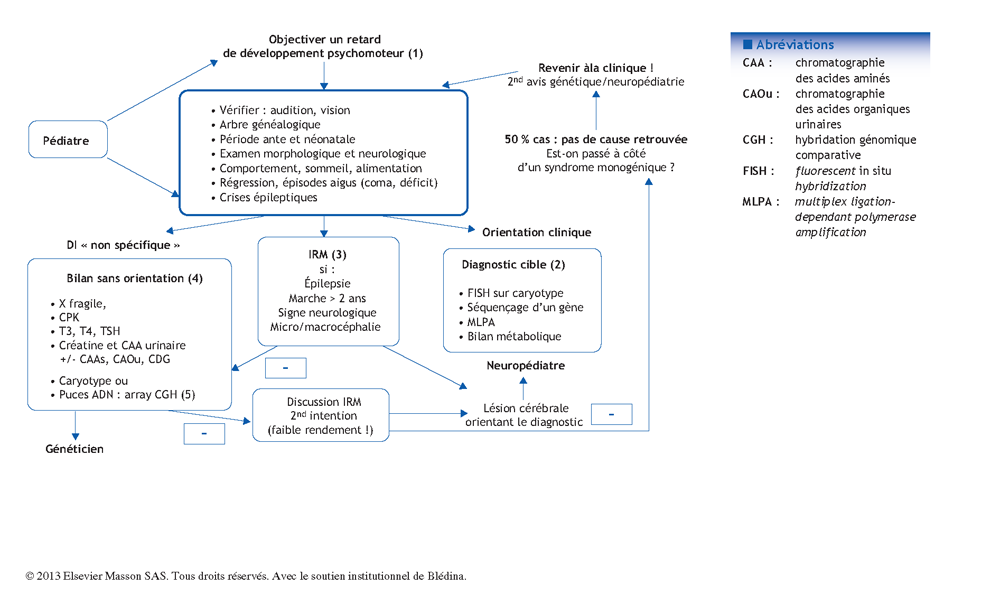
Arbre décisionnel – Commentaires
Deux à trois pour cent des nouveau- nés auront une déficience intellectuelle. En faire le diagnostic pour mettre en œuvre la prise en charge, mais aussi en rechercher la cause font partie des missions du pédiatre. Le schéma d’investigation étiologique proposé ici a été validé par le groupe de travail du réseau DéfiScience**. L’objectif principal d’un diagnostic étiologique reste avant tout la recherche d’une cause curable et/ou possiblement récurrente impliquant un conseil génétique fiable. Le choix des examens doit rester avant tout clinique. Mais, très souvent, la déficience intellectuelle est isolée, sans orientation diagnostique. Cette partie du diagnostic peut nécessiter de recourir à un avis spécialisé en génétique et ou neuropédiatrie. Cependant, le pédiatre se doit de connaître sans rentrer dans les détails, les indications de l’imagerie et des principaux examens biologiques, et avoir entendu parler de « puce à ADN », maintenant disponible en routine, pour pouvoir répondre aux questions des parents.
(1) Du retard psychomoteur à la déficience intellectuelle
Plus le handicap est sévère, plus les signes d’appel seront précoces : hypotonie néonatale, retard postural, mauvaise interaction précoce pour une déficience sévère, retard massif du langage, agitation psychomotrice, pour une déficience moyenne, trouble des apprentissages scolaire en primaire pour une déficience légère. Chaque symptôme d’appel amène des diagnostics différentiels multiples, neurologiques, sensoriels, psychiatriques, somatiques, que l’on ne peut synthétiser ici. Les échelles de développement telles que la Brunet Lézine sont utiles pour évaluer précisément en consultation les quatre domaines de développement : posture, coordination motrice fine, langage et interaction sociale. Plus l’enfant est jeune, plus le pronostic cognitif est délicat et la part de déficience sera appréciée selon la manière dont l’enfant mobilise ses compétences et « rattrape » son retard, avec une stimulation bien conduite (kinésithérapeute, puis psychomotricien, prise en charge CAMSP ou CMP). Pour les déficiences légères et moyennes, un test psychométrique est indispensable pour confirmer les difficultés de raisonnement et différentier d’un trouble spécifique des apprentissages.
Si l’enfant ne rattrape pas son retard, une cause (infectieuse, toxique, génétique) doit être recherchée sans attendre, en évitant de rassurer à tort des parents qui ont bien perçu ce décalage comme anormal, et sans mettre les symptômes sur le compte de facteurs purement psychologiques. Il ne faut certes pas inquiéter inutilement, mais surtout savoir enclencher des explorations étiologiques à temps pour répondre aux quatre questions des parents : pourquoi a- t-il un retard ? Comment va- t-il évoluer ? Y a- t-il un traitement ? Quel risque pour un autre enfant ?
(2) Diagnostic ciblé selon la clinique
La plupart des syndromes cliniques peuvent aujourd’hui être confirmés par un test génétique. Cela peut être le séquençage d’un gène ou d’un groupe de gènes responsables d’un syndrome clinique, ou bien l’hybridation sur caryotype d’une sonde spécifique (FISH, fluorescent in situ hybridization) qui permet d’identifier sur un caryotype des microdélétions, sur la région chromosomique correspondant à un syndrome suspecté cliniquement (Williams, DiGeorge, Smith- Magenis…). Des techniques de génétique moléculaire quantitative, telle que la mlPA (multiplex ligation- dependant polymerase amplification) permettent de dénombrer le nombre de copies d’une région chromosomique donnée, soit pour identifier une délétion/duplication d’un gène cible évoqué cliniquement mais dont la séquence est apparemment normale, ou pour étudier d’un coup plusieurs dizaines de loci, dont, par exemple les régions subtélomériques, sièges de 2,5 % à 5 % d’anomalies non visibles sur le caryotype. Le groupe très vaste des maladies héréditaires du métabolisme, sera évoqué devant certaines situations cliniques : atteinte multiviscérale ou sensorielle, signes neurologiques progressifs ou fluctuants, épisodes neurologiques aigus inexpliqués,
intolérances aux protéines, justifient ces explorations spécialisées. En l’absence d’orientation clinique, une chromatographie des acides aminés sanguins (CAAs), une chromatographie des acides organiques urinaires (CAOu) et un CDG (trouble de N- glycosylation des protéines) seront demandés en cas de consanguinité des parents ou de déficience intellectuelle sévère ou profonde. Par ailleurs, en l’absence d’IRM avec spectroscopie, un dosage urinaire du guanidinoacétate et de la créatine, permet de rechercher un défaut de synthèse ou de transport de la créatine, peu spécifique cliniquement.
(3) L’imagerie cérébrale par résonance magnétique (IRM)
L’IRM serait anormale dans 30 % des cas de RM, dans plus de 40 % des examens lorsqu’il y a une symptomatologie neurologique, mais dans moins de 15 % quand il n’y a pas de signe d’appel. Toutefois, les anomalies observées ne fournissent la clé du diagnostic étiologique que dans 2 à 4 % des cas. Une IRM est indiquée en cas de macro- ou de microcéphalie, en cas d’épilepsie ou de régression, de signes neurologiques, en cas de retard sévère avec retard moteur (marche non acquise à 2 ans). En dehors de ces situations, le rendement est quasi nul et l’IRM ne sera pas faite en première intention.
(4) Bilan sans orientation clinique
Le syndrome X fragile est une pathologie fréquente (1/5 000 garçons et 1/9 000 filles) dont le diagnostic clinique est parfois difficile. Son caractère familial et ses conséquences pour le conseil génétique justifient sa réalisation systématique devant toute DI non syndromique. Chez un garçon, le dosage des CPK dépiste précocement une myopathie de Duchenne, qui peut se manifester d’abord par un retard psychomoteur. Le bilan thyroïdien est indispensable, car de nombreux syndromes s’accompagnent d’une dysthyroïdie source de surhandicap. De plus, la comparaison de T3 et T4 permet de suspecter un déficit en transporteur cérébral de la T3 (gène MCT8), affection liée à l’X, caractérisée par des taux élevés de T3, contrastant avec des valeurs de T4 basse.
(5) Évolution des techniques de génétique « pangénomiques », sans orientation clinique
Le caryotype est un examen pangénomique de faible sensibilité. En dehors de la trisomie 21 (qui explique à elle seule environ 10 % de l’ensemble des DI), le rendement diagnostique du caryotype est faible. L’hybridation génomique comparative (CGH : comparative genomic hybridization) sur matrice ordonnée (microréseau ou array CGH ou « puce à ADN ») constitue une véritable révolution diagnostique. La puissance de la technique vient de sa capacité à analyser d’un seul coup un très grand nombre de loci. Dans une population d’enfants présentant une déficience intellectuelle à caryotype normal, 10 à 20 % présentent des remaniements génomiques significatifs en CGH. Certaines variations observées sont de simples polymorphismes ; d’autres peuvent révéler des facteurs de risque non recherchés tels que prédisposition à des cancers. La complexité de l’information recueillie doit être impérativement expliquée aux parents lors de la signature du consentement au test génétique. Avec ces précautions, il est désormais recommandé d’abandonner le caryotype au profit d’une puce faite en première intention, puisque plus de 80 % des anomalies chromosomiques sont infra- cytogénétiques.
Liens d’intérêts
Les auteurs ont déclaré n’avoir aucun conflit d’intérêts pour cet article.
Références
Decobert F, Grabar S, Merzoug V, et al. Unexplained mental retardation: is brain MRI useful? Pediatr Radiol 2005;35:587-96.
McDonald L, Rennie A, Tolmie J, et al. Investigation of global developmental delay. Arch Dis Child 2006;91:701-5.
Sagoo GS, Butterworth AS, Sanderson S, et al. Array CGH in patients with learning disability (mental retardation) and congenital anomalies: updated systematic review and meta- analysis of 19 studies and 13,926 subjects. Genet Med 2009;11:139-46.
Verloes A, Héron D, Billette de Villemeur T, et al. Stratégie d’exploration d’une déficience intellectuelle inexpliquée. Arch Pediatr2012;19:194-207.