
Vous êtes ici
Thématique:
L'enfant trop petit
R. Coutant a, M. Nicolinob
aUnité d’endocrinologie-diabétologie pédiatrique, pôle enfant, CHU Angers, 4 rue Larrey, 49000 Angers, France bService d’endocrinologie-diabétologie pédiatrique, hôpital Debrousse, 29 rue Sœur-Bouvier, 69000 Lyon, France Auteur correspondant - Adresse e-mail : recoutant@chu-angers.fr (R. Coutant).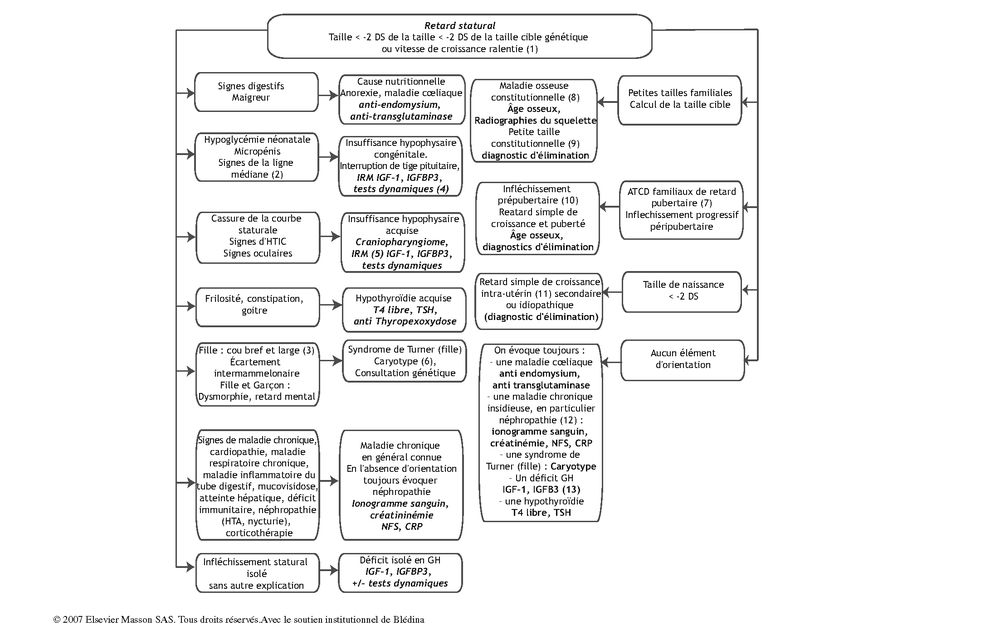
Arbre décisionnel – Commentaires
(1) Le retard statural est défini par une taille < -2 DS pour l’âge et le sexe (courbes françaises de Sempé et Pédron), une taille < -2 DS de la taille cible parentale (taille cible garçon = (T père + T mère + 13)/2 ; taille cible fille = (T père + T mère – 13)/2), ou une vitesse de croissance faible (la taille ne reste pas sur une DS stable).
(2) Les insuffisances hypophysaires congénitales sont suspectées devant une histoire d’hypoglycémie néonatale, un micro pénis (présent chez 50 % des garçons atteints), des signes évoquant une atteinte de la ligne médiane (colobome rétinien ou irien, fente labiale ou labio-palatine…).
(3) Les signes cliniques du syndrome de Turner peuvent être discrets voire absents. Les retards staturaux associées à une dysmorphie, d’autres malformations (cardiaques…) ou un retard mental évoquent une petite taille syndromique (exemple : syndrome de Noonan) nécessitant une consultation de génétique.
(4) Lorsque l’on suspecte une insuffisance hypophysaire congénitale, les mesures d’IGF-1 et IGFB-3 orientent vers un déficit en GH, de T4 libre et TSH vers une insuffisance thyréotrope. La réalisation de tests dynamiques est utile pour apprécier l’ensemble des fonctions hypophysaires. L’IRM hypothalamo-hypophysaire est indispensable : l’interruption de la tige pituitaire est la première cause d’insuffisance hypophysaire congénitale.
(5) L’IRM est l’examen le plus important devant une cassure de la taille, à la recherche d’une tumeur de la région hypothalamo-hypophysaire. Le diagnostic le plus fréquent est le craniopharyngiome. En dehors des tumeurs, les retards staturaux d’origine psycho-sociale sont la seconde cause de cassure de la taille.
(6) Le caryotype est un examen nécessaire chez toutes les filles de petite taille.
(7) La connaissance de l’âge de la ménarche de la mère, d’une notion de croissance tardive chez le père, permet d’estimer les ATCD familiaux de retard pubertaire.
(8) Les maladies osseuses constitutionnelles correspondent à des maladies monogéniques affectant des gènes impliqués dans la croissance et le modelage de l’os et du cartilage. Les plus fréquentes sont la dyschondrostéose, l’hypochondroplasie, la pseudohypoparathyroïdie, et les dysplasies poly-épiphysaires. Les radiographies squelettiques de dépistage comprennent un bassin F, un rachis lombaire F et P, un avant bras F et P, et un âge osseux (main + poignet G de face).
(9) Un arbre généalogique de petites tailles peut correspondre à un diagnostic de petite taille familiale (hérédité polygénique, pas de maladie monogénique), mais aussi à une maladie osseuse constitutionnelle (de transmission dominante le plus souvent).
(10) L’infléchissement prépubertaire et le retard simple de croissance et de puberté sont les causes les plus fréquentes d’infléchissement statural à l’adolescence. C’est un diagnostic présomptif, retenu en l’absence d’autres signes, lorsqu’il existe des ATCD familiaux analogues, lorsque l’âge osseux est < 11 ans chez la fille et < 13 ans chez le garçon, et lorsque l’infléchissement est modéré (perte staturale de moins de 1 DS). L’enfant doit être suivi jusqu’à l’observation de l’accélération staturale contemporaine de la puberté.
(11) Quatre-vingt-cinq pour cent des enfants nés petits pour l’âge gestationnel rattrapent une taille > -2 DS avant l’âge de 4 ans. Ceux qui ne rattrapent pas peuvent être « idiopathiques », mais aussi correspondre à des syndromes (Silver-Russel, Turner, Noonan, Williams), ou à d’authentiques maladies gênant la croissance (déficit en GH, dysplasie squelettique…). Une investigation diagnostique est nécessaire.
(12) Les maladies chroniques sont en général symptomatiques. Les plus « silencieuses » sont les maladies rénales et la maladie cœliaque. On les évoque toujours.
(13) Les taux sanguins d’IGF-1 et IGFBP-3 sont des indicateurs de la sécrétion d’hormone de croissance. Les kits de dosage ne sont pas encore standardisés, ce qui pose des problèmes de fiabilité. Il faut utiliser ces mesures comme un élément d’orientation, et s’interroger sur un déficit en GH lorsque les valeurs sont inférieures à la moyenne pour l’âge et le sexe. Dans ce cas, un test dynamique de stimulation de la GH peut être discuté.
Références
New Maria. Growth and growth disorders. Available from URL: http://www.endotext.org/pediatrics/index.htm
Bourrillon A. Diagnostic d’un retard de croissance staturo-pondérale. In Pédiatrie. A Bourrillon. Masson, Paris 2005:223-35.